現在7歲大的男童「柴柴」,在一出生僅17天就確診罹患罕見結節硬化症與多重障礙疾病,因此遭受父母虐待,多次進出住院,收治他的中山醫學大學附設醫院新生兒科主任王杏安,在治療過程中被柴柴的笑容融化,雖單身未婚卻下定決心收養柴柴,歷經3年的繁瑣程序,終於在去年成為合法母子。

現在7歲大的男童「柴柴」,在一出生僅17天就確診罹患罕見結節硬化症與多重障礙疾病,因此遭受父母虐待,多次進出住院,收治他的中山醫學大學附設醫院新生兒科主任王杏安,在治療過程中被柴柴的笑容融化,雖單身未婚卻下定決心收養柴柴,歷經3年的繁瑣程序,終於在去年成為合法母子。

孩子一出生,先買好保險才能做新生兒自費檢查嗎?有等待期嗎?台北榮總近期收治一名王姓男嬰,出生後27天出現明顯呼吸困難症狀,緊急就醫發現心臟肥大、肌肉酵素增高,高度懷疑為龐貝氏症,需馬上插管治療,醫師詢問後才知道,爸媽原本預計嬰兒滿月納保後,才進行新生兒篩檢自費項目,僅幾天之差導致王小弟無法及時篩檢出龐貝氏症,險害孩子一生而悔不當初。

10歲的小晴在出生後4個月大時,被發現不會翻身及抬頭,經確診為先天性罕病SMA第1型。1歲時因感染肺炎導致呼吸衰竭,入住加護病房,被醫師判定生命僅剩數月,但家屬不放棄治療,隨時陪伴拍痰、抽痰,小晴每天需配戴呼吸器23個小時;4歲多開始用藥後,居家呼吸器縮減為每天使用10小時,抽痰1至2次,她從原本無法講完整句子到現在能唱完一首兒歌。

隨時都要擔心肚子痛到像生產痛,甚至喉嚨腫痛無法呼吸,當心可能罹患罕病「遺傳性血管性水腫」(HAE)!一名35歲壯男突然休克昏倒,送醫診治意外發現自己為HAE患者,天天擔心發病像不定時炸彈。過敏免疫風濕科醫師提醒,此病經常與過敏、蕁麻疹混淆,但若未及時治療恐致命,可透過3大症狀特點來分辨。

不明手麻、腳痛竟是心肌病變!70歲的邵小姐在4年前開始感到手指麻木、無力甚至間歇疼痛,就醫後被診斷為板機指與腕隧道症候群,且出現越來越虛弱、水腫等症狀,不僅連洗澡都氣喘吁吁,體重也急遽下降,反覆進行手術卻治療無果。因弟弟也有類似症狀,才在親友建議下去做基因檢測,最後確診為罕見的遺傳型心肌病變(ATTR-CM)。
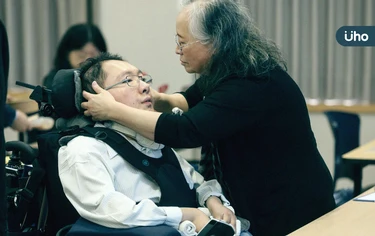
脫口秀節目《賀瓏夜夜秀》日前邀請中國媒體人王志安評論台灣選舉,卻因模仿嘲諷身障律師陳俊翰引發撻伐,引發社會關注,主持人賀瓏昨日終於在節目中公開致歉。人權律師陳俊翰身為罕病「脊髓性肌肉萎縮症」(SMA)病友,指出全台仍有300多位患者無法使用藥物,自己也處於「望藥興嘆」困境。

台中69歲黃女士10多年來,一直因頭暈問題困擾不已,曾經至台中各大醫院耳鼻喉科、神經內科和心臟科求診,始終未能找到頭暈的根本原因。近日到仁愛長庚合作聯盟醫院耳鼻喉科就診,醫師發現她長期流鼻血,並在下唇發現一些類似血管瘤的小突起,內視鏡檢查有出血現象,加上眼白非常白,走進診間時明顯無力、呼吸急促,懷疑她可能患有「遺傳性出血性血管增生症」。

許多癌症、罕病病友,常因等不到新藥納入健保給付,只能咬牙自費大筆金錢苦撐。根據統計,癌症新藥平均審查等待時間約需783天(約26.1個月),不僅與健保署曾對外公布的411天(約13.7個月)落差甚遠,病人及家屬也無從得知藥品審查進度。鑑此,癌症希望基金會、台灣病友聯盟及罕見疾病基金會共同倡議,健保新藥審查時程應符合「透明化、可被追蹤」2大訴求。

