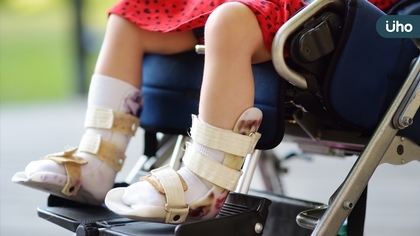
「罕病生命鬥士」陳俊翰疑似感冒引起併發症,2月11日凌晨不幸逝世,享年40歲。陳俊翰因罹患罕見疾病——脊髓性肌肉萎縮症(SMA),漸漸失去行動力,靠嘴巴控制輪椅和寫字,取得美國法學博士與律師執照,卻捨棄海外治療機會,返台爭取罕病平等醫療。陳俊翰生前出席公開活動時感慨SMA藥物問世,台灣有條件的給付,讓病人從滿心希望藥物治療,到「有藥可用,但不包括我」的沮喪,等待的過程與結果相當殘忍與煎熬。 SMA運動神經元退化的遺傳性疾病 臺北榮民總醫院神經醫學中心周邊神經科蕭丞宗醫師表示,SMA是一種因運動神經元退化,導致肌肉無力及萎縮的體染色體隱性遺傳疾病,使得運動能力下降、肌肉萎縮、最後可能無法行走,甚至影響呼吸肌與吞嚥功能,導致死亡。 SMA依發病年齡與最佳運動功能分4種治療類型:第一型,約6成SMA病友在出生6個月內發病,若未能及早治療,2歲前死亡率高達8成。第二型,6個月到18個月間發病,獨坐能力逐漸退化,最後只能臥床。第三型,18個月至18歲間發病,初期可行走,但漸失去行走能力,最後輪椅代步。第四型。成人輕微運動功能障礙。 目前SMA有背針、口服、基因治療等3種治療方式,對病人的肌肉萎縮症狀有明顯的改善。台灣的SMA治療跟國外相比有很大落差,許多國家對SMA治療採取全給付政策,只要有確定診斷,不受年紀跟嚴重程度的限制;台灣則有諸多限制,例如,超過7歲的病人,需具備3歲前病史及上肢運動功能RULM≧15,很多SMA病人不容易達到這個門檻。 4900萬元基因治療藥 健保僅限6個月以下發病且帶基因突變 要價達4900萬元的SMA基因治療,健保給付限制6個月以下發病且帶有基因突變者,預估一年8名病童受惠。但目前仍有300多位SMA患者無法符合健保給付資格,需要背針與口服治療,他們陷在「有藥可用,但不包括我」的沮喪中,極需集社會眾人之力,照顧他們的需要。 診間一位48歲的男士,一兩歲左右發病,媽媽細數他的成長過程,他的運動功能比同齡者來得慢,兩歲多學會走路但經常跌倒,幼兒到國中階段運動功能明顯不如其他同學,30歲左右漸漸需要輪椅代步。所幸,目前已治療八個月,運動功能明顯進步,除了在床上可以延長把腿抬高的時間,原本沒辦法做趴的姿勢,現在可以,從原本只能走30公尺現在可走到快60公尺。 蕭丞宗提醒,每一位罕病病友都在跟時間賽跑,急需要藥物阻止疾病惡化,臨床經驗指出,病人如果可以及早接受治療,無論兒童與成人,都有機會延緩惡化甚至讓運動功能變進步,也能降低感染引發肺炎等嚴重併發症的風險。 (圖/「罕病生命鬥士」陳俊翰律師)